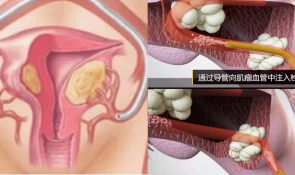
IntelligentOncology以智能肿瘤学科为核心主题,致力于发布高质量的原创性研究、综述和评论,通过汇集这一多学科领域最重要的进展,促进临床肿瘤学、信息科学、数学等多学科融合的理论与应用研究。稿源方向包括肿瘤人工智能技术的基础理论与临床肿瘤学研究应用、智能肿瘤学临床试验、药物研发等。包括医学、药学、工学和理学在肿瘤诊疗中的相关研究。本刊具有广泛的选稿和组稿范围,为肿瘤人工智能相关理论研究和临床诊疗应用研究提供成果分享和总结交流平台。本刊的创办团队汇聚了临床肿瘤学与人工智能领域全球优秀的科学家,他们将智能肿瘤学推向全球领先地位,引领智能肿瘤学的前沿学科传播。《智能肿瘤学》融合了人工智能和临床肿瘤学的元素,重庆大学附属肿瘤医院院长徐波教授为本刊创刊主编,哈佛大学医学院和麻省总医院的蔡文立教授及香港大学孔凤鸣教授为本刊联合主编。本刊编辑委员会由40名知名专家组成,其中包括医生、科学家和工程师。